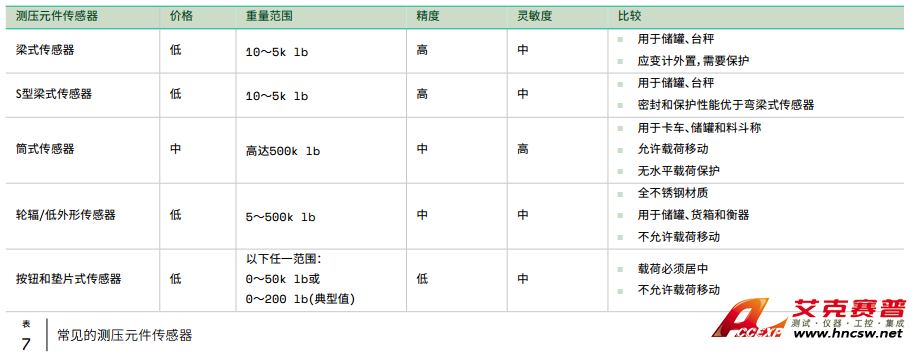
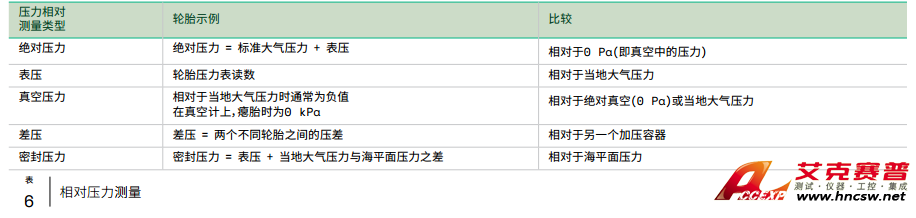
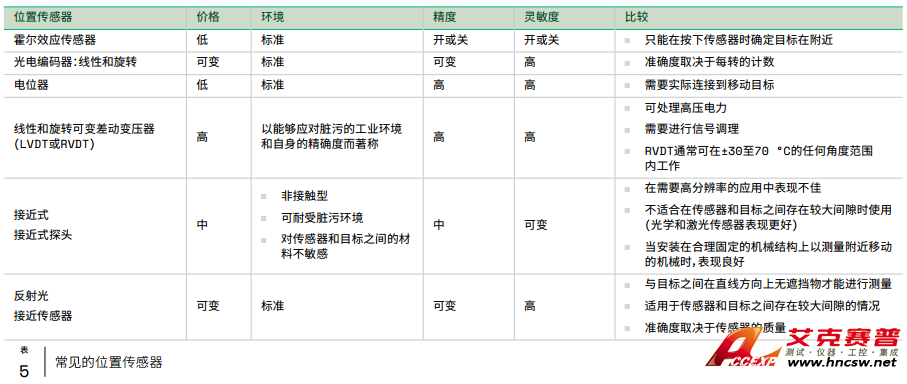
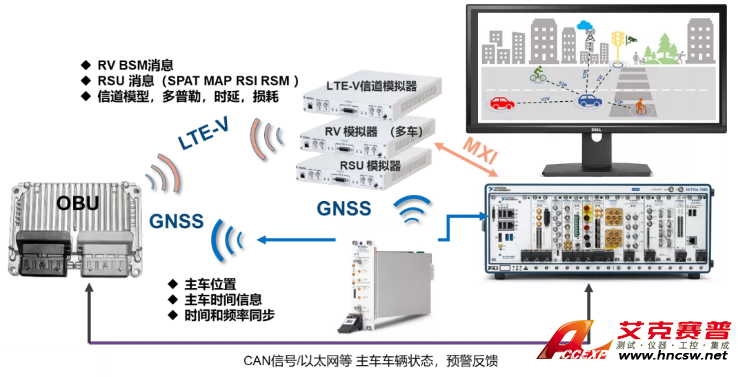
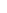分光光度计的简单描述

2013-08-19
二分光光度计的简单原理
分光光度计采用一个可以产生多个波长的光源,通过系列分光装置,从而产生特定波长的光源,光源透过测试的样品后,部分光源被吸收,计算样品的吸光值,从而转化成样品的浓度。样品的吸光值与样品的浓度成正比。
核酸的定量
核酸的定量是分光光度计使用频率最高的功能。可以定量溶于缓冲液的寡核苷酸,单链、双链DNA,以及RNA。核酸的最高吸收峰的吸收波长260 nm。 每种核酸的分子构成不一,因此其换算系数不同。定量不同类型的核酸,事先要选择对应的系数。如:1OD的吸光值分别相当于50μg/ml的dsDNA,37μg/ml的ssDNA,40μg/ml的RNA,30μg/ml的Olig。测试后的吸光值经过上述系数的换算,从而得出相应的样品浓度。测试前,选择正确的程序,输入原液和稀释液的体积,尔后测试空白液和样品液。然而,实验并非一帆风顺。读数不稳定可能是实验者最头痛的问题。灵敏度越高的仪器,表现出的吸光值漂移越大。
事实上,分光光度计的设计原理和工作原理,允许吸光值在一定范围内变化,即仪器有一定的准确度和精确度。如EppendorfBiophotometer的准确度≤1.0%(1A)。这样多次测试的结果在均值1.0%左右之间变动,都是正常的。另外,还需考虑核酸本身物化性质和溶解核酸的缓冲液的pH值,离子浓度等:在测试时,离子浓度太高,也会导致读数漂移,因此建议使用pH值一定、离子浓度较低的缓冲液,如TE,可大大稳定读数。样品的稀释浓度同样是不可忽视的因素:由于样品中不可避免存在一些细小的颗粒,尤其是核酸样品。这些小颗粒的存在干扰测试效果。为了最大程度减少颗粒对测试结果的影响,要求核酸吸光值至少大于0.1A,吸光值最好在0.1-1.5A。在此范围内,颗粒的干扰相对较小,结果稳定。
从而意味着样品的浓度不能过低,或者过高(超过光度计的测试范围)。最后是操作因素,如混合要充分,否则吸光值太低,甚至出现负值;混合液不能存在气泡,空白液无悬浮物,否则读数漂移剧烈;必须使用相同的比色杯测试空白液和样品,否则浓度差异太大;换算系数和样品浓度单位选择一致;不能采用窗口磨损的比色杯;样品的体积必须达到比色杯要求的最小体积等多个操作事项。
除了核酸浓度,分光光度计同时显示几个非常重要的比值表示样品的纯度,如 A 260 / A 280的比值,用于评估样品的纯度,因为蛋白的吸收峰是 280 nm。纯净的样品,比值大于 1.8(DNA)或者2.0(RNA)。如果比值低于 1.8 或者2.0,表示存在蛋白质或者酚类物质的影响。A 230表示样品中存在一些污染物,如碳水化合物,多肽,苯酚等,较纯净的核酸 A 260 / A 230 的比值大于 2.0。A 320检测溶液的混浊度和其他干扰因子。纯样品,A 320 一般是 0。
蛋白质的直接定量(UV法)
这种方法是在280 nm波长,直接测试蛋白。选择 Warburg公式,光度计可以直接显示出样品的浓度,或者是选择相应的换算方法,将吸光值转换为样品浓度。
蛋白质测定过程非常简单,先测试空白液,然后直接测试蛋白质。由于缓冲液中存在一些杂质,一般要消除320 nm的“背景”信息,设定此功能“开”。与测试核酸类似,要求 A 280 的吸光值至少大于 0.1A,最佳的线性范围在1.0-1.5 之间。实验中选择 Warburg 公式显示样品浓度时,发现读数“漂移”。这是一个正常的现象。事实上,只要观察A 280的吸光值的变化范围不超过 1%,表明结果非常稳定。
漂移的原因是因为 Warburg公式吸光值换算成浓度,乘以一定的系数,只要吸光值有少许改变,浓度就会被放大,从而显得结果很不稳定。
蛋白质直接定量方法,适合测试较纯净、成分相对单一的蛋白质。紫外直接定量法相对于比色法来说,速度快,操作简单;但是容易受到平行物质的干扰,如DNA 的干扰;另外敏感度低,要求蛋白的浓度较高。
分光光度计采用一个可以产生多个波长的光源,通过系列分光装置,从而产生特定波长的光源,光源透过测试的样品后,部分光源被吸收,计算样品的吸光值,从而转化成样品的浓度。样品的吸光值与样品的浓度成正比。
核酸的定量
核酸的定量是分光光度计使用频率最高的功能。可以定量溶于缓冲液的寡核苷酸,单链、双链DNA,以及RNA。核酸的最高吸收峰的吸收波长260 nm。 每种核酸的分子构成不一,因此其换算系数不同。定量不同类型的核酸,事先要选择对应的系数。如:1OD的吸光值分别相当于50μg/ml的dsDNA,37μg/ml的ssDNA,40μg/ml的RNA,30μg/ml的Olig。测试后的吸光值经过上述系数的换算,从而得出相应的样品浓度。测试前,选择正确的程序,输入原液和稀释液的体积,尔后测试空白液和样品液。然而,实验并非一帆风顺。读数不稳定可能是实验者最头痛的问题。灵敏度越高的仪器,表现出的吸光值漂移越大。
事实上,分光光度计的设计原理和工作原理,允许吸光值在一定范围内变化,即仪器有一定的准确度和精确度。如EppendorfBiophotometer的准确度≤1.0%(1A)。这样多次测试的结果在均值1.0%左右之间变动,都是正常的。另外,还需考虑核酸本身物化性质和溶解核酸的缓冲液的pH值,离子浓度等:在测试时,离子浓度太高,也会导致读数漂移,因此建议使用pH值一定、离子浓度较低的缓冲液,如TE,可大大稳定读数。样品的稀释浓度同样是不可忽视的因素:由于样品中不可避免存在一些细小的颗粒,尤其是核酸样品。这些小颗粒的存在干扰测试效果。为了最大程度减少颗粒对测试结果的影响,要求核酸吸光值至少大于0.1A,吸光值最好在0.1-1.5A。在此范围内,颗粒的干扰相对较小,结果稳定。
从而意味着样品的浓度不能过低,或者过高(超过光度计的测试范围)。最后是操作因素,如混合要充分,否则吸光值太低,甚至出现负值;混合液不能存在气泡,空白液无悬浮物,否则读数漂移剧烈;必须使用相同的比色杯测试空白液和样品,否则浓度差异太大;换算系数和样品浓度单位选择一致;不能采用窗口磨损的比色杯;样品的体积必须达到比色杯要求的最小体积等多个操作事项。
除了核酸浓度,分光光度计同时显示几个非常重要的比值表示样品的纯度,如 A 260 / A 280的比值,用于评估样品的纯度,因为蛋白的吸收峰是 280 nm。纯净的样品,比值大于 1.8(DNA)或者2.0(RNA)。如果比值低于 1.8 或者2.0,表示存在蛋白质或者酚类物质的影响。A 230表示样品中存在一些污染物,如碳水化合物,多肽,苯酚等,较纯净的核酸 A 260 / A 230 的比值大于 2.0。A 320检测溶液的混浊度和其他干扰因子。纯样品,A 320 一般是 0。
蛋白质的直接定量(UV法)
这种方法是在280 nm波长,直接测试蛋白。选择 Warburg公式,光度计可以直接显示出样品的浓度,或者是选择相应的换算方法,将吸光值转换为样品浓度。
蛋白质测定过程非常简单,先测试空白液,然后直接测试蛋白质。由于缓冲液中存在一些杂质,一般要消除320 nm的“背景”信息,设定此功能“开”。与测试核酸类似,要求 A 280 的吸光值至少大于 0.1A,最佳的线性范围在1.0-1.5 之间。实验中选择 Warburg 公式显示样品浓度时,发现读数“漂移”。这是一个正常的现象。事实上,只要观察A 280的吸光值的变化范围不超过 1%,表明结果非常稳定。
漂移的原因是因为 Warburg公式吸光值换算成浓度,乘以一定的系数,只要吸光值有少许改变,浓度就会被放大,从而显得结果很不稳定。
蛋白质直接定量方法,适合测试较纯净、成分相对单一的蛋白质。紫外直接定量法相对于比色法来说,速度快,操作简单;但是容易受到平行物质的干扰,如DNA 的干扰;另外敏感度低,要求蛋白的浓度较高。